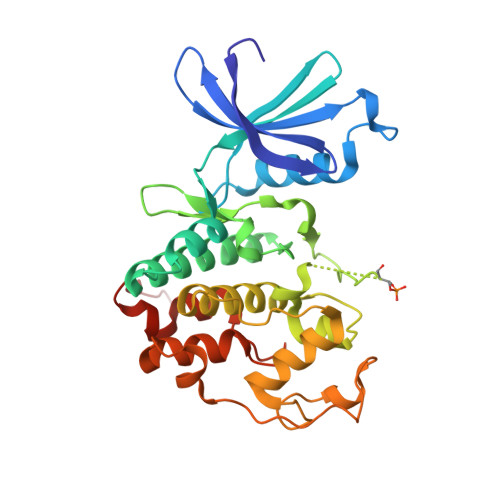
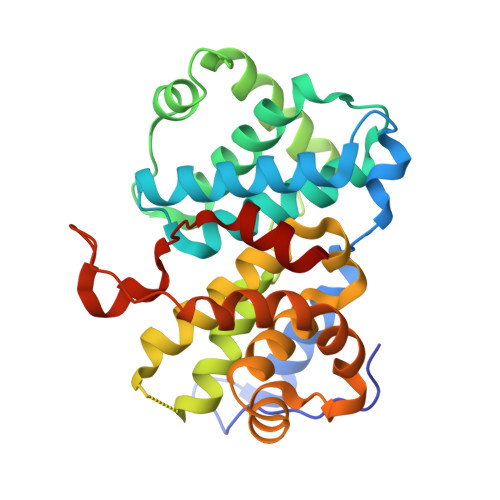
An allosteric cyclin E-CDK2 site mapped by paralog hopping with covalent probes.
Zhang, Y., Liu, Z., Hirschi, M., Brodsky, O., Johnson, E., Won, S.J., Nagata, A., Bezwada, D., Petroski, M.D., Majmudar, J.D., Niessen, S., VanArsdale, T., Gilbert, A.M., Hayward, M.M., Stewart, A.E., Nager, A.R., Melillo, B., Cravatt, B.F.(2025) Nat Chem Biol 21: 420-431
- PubMed: 39294320 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1038/s41589-024-01738-7
- Primary Citation Related Structures:
8VQ3, 8VQ4 - PubMed Abstract:
More than half of the ~20,000 protein-encoding human genes have paralogs. Chemical proteomics has uncovered many electrophile-sensitive cysteines that are exclusive to subsets of paralogous proteins. Here we explore whether such covalent compound-cysteine interactions can be used to discover ligandable pockets in paralogs lacking the cysteine. Leveraging the covalent ligandability of C109 in the cyclin CCNE2, we substituted the corresponding residue in paralog CCNE1 to cysteine (N112C) and found through activity-based protein profiling that this mutant reacts stereoselectively and site-specifically with tryptoline acrylamides. We then converted the tryptoline acrylamide-CCNE1-N112C interaction into in vitro NanoBRET (bioluminescence resonance energy transfer) and in cellulo activity-based protein profiling assays capable of identifying compounds that reversibly inhibit both the N112C mutant and wild-type CCNE1:CDK2 (cyclin-dependent kinase 2) complexes. X-ray crystallography revealed a cryptic allosteric pocket at the CCNE1:CDK2 interface adjacent to N112 that binds the reversible inhibitors. Our findings, thus, show how electrophile-cysteine interactions mapped by chemical proteomics can extend the understanding of protein ligandability beyond covalent chemistry.
- Department of Chemistry, The Scripps Research Institute, La Jolla, CA, USA.
Organizational Affiliation: