Millisecond cryo-trapping by the spitrobot crystal plunger simplifies time-resolved crystallography.
Mehrabi, P., Sung, S., von Stetten, D., Prester, A., Hatton, C.E., Kleine-Dopke, S., Berkes, A., Gore, G., Leimkohl, J.P., Schikora, H., Kollewe, M., Rohde, H., Wilmanns, M., Tellkamp, F., Schulz, E.C.(2023) Nat Commun 14: 2365-2365
- PubMed: 37185266 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1038/s41467-023-37834-w
- Primary Citation Related Structures:
8AW8, 8AW9, 8AWB, 8AWC, 8AWD, 8AWE, 8AWF, 8AWS, 8AWU, 8AWV, 8AWX, 8AWY, 8B03, 8B05, 8B06, 8B08, 8B2O, 8B2V, 8B2W, 8B3M - PubMed Abstract:
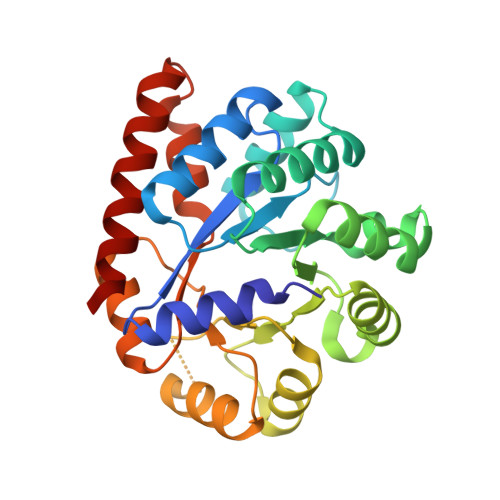
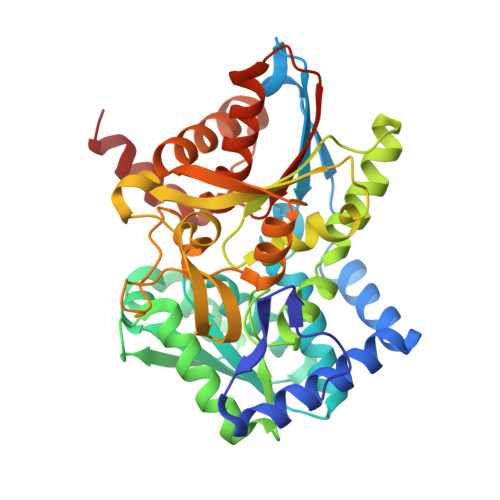
We introduce the spitrobot, a protein crystal plunger, enabling reaction quenching via cryo-trapping with a time-resolution in the millisecond range. Protein crystals are mounted on canonical micromeshes on an electropneumatic piston, where the crystals are kept in a humidity and temperature-controlled environment, then reactions are initiated via the liquid application method (LAMA) and plunging into liquid nitrogen is initiated after an electronically set delay time to cryo-trap intermediate states. High-magnification images are automatically recorded before and after droplet deposition, prior to plunging. The SPINE-standard sample holder is directly plunged into a storage puck, enabling compatibility with high-throughput infrastructure. Here we demonstrate binding of glucose and 2,3-butanediol in microcrystals of xylose isomerase, and of avibactam and ampicillin in microcrystals of the extended spectrum beta-lactamase CTX-M-14. We also trap reaction intermediates and conformational changes in macroscopic crystals of tryptophan synthase to demonstrate that the spitrobot enables insight into catalytic events.
- Institute for Nanostructure and Solid-State Physics, Universität Hamburg, Hamburg, Germany. pedram.mehrabi@uni-hamburg.de.
Organizational Affiliation: