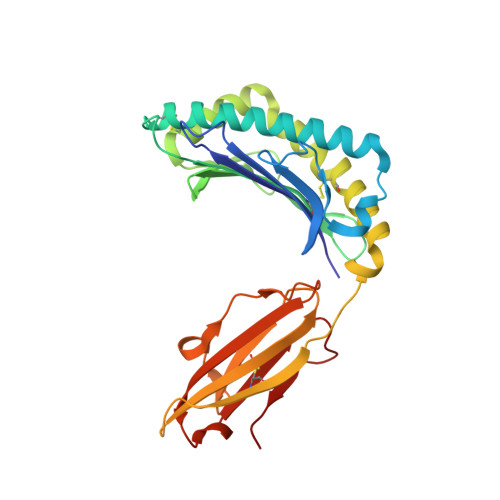
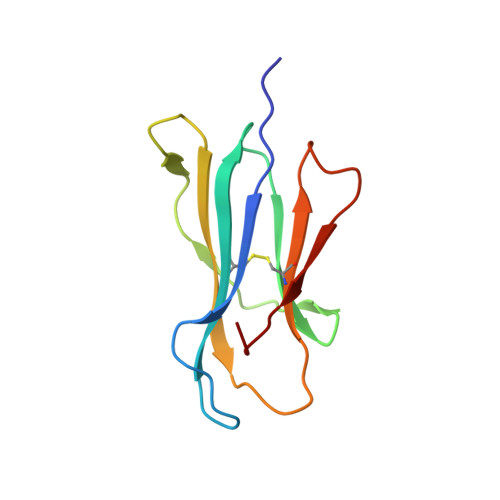
Structures of peptide-free and partially loaded MHC class I molecules reveal mechanisms of peptide selection.
Anjanappa, R., Garcia-Alai, M., Kopicki, J.D., Lockhauserbaumer, J., Aboelmagd, M., Hinrichs, J., Nemtanu, I.M., Uetrecht, C., Zacharias, M., Springer, S., Meijers, R.(2020) Nat Commun 11: 1314-1314
- PubMed: 32161266 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1038/s41467-020-14862-4
- Primary Citation Related Structures:
6TDO, 6TDP, 6TDQ, 6TDR, 6TDS - PubMed Abstract:
Major Histocompatibility Complex (MHC) class I molecules selectively bind peptides for presentation to cytotoxic T cells. The peptide-free state of these molecules is not well understood. Here, we characterize a disulfide-stabilized version of the human class I molecule HLA-A*02:01 that is stable in the absence of peptide and can readily exchange cognate peptides. We present X-ray crystal structures of the peptide-free state of HLA-A*02:01, together with structures that have dipeptides bound in the A and F pockets. These structural snapshots reveal that the amino acid side chains lining the binding pockets switch in a coordinated fashion between a peptide-free unlocked state and a peptide-bound locked state. Molecular dynamics simulations suggest that the opening and closing of the F pocket affects peptide ligand conformations in adjacent binding pockets. We propose that peptide binding is co-determined by synergy between the binding pockets of the MHC molecule.
- Department of Life Sciences and Chemistry, Jacobs University Bremen, Bremen, Germany.
Organizational Affiliation: