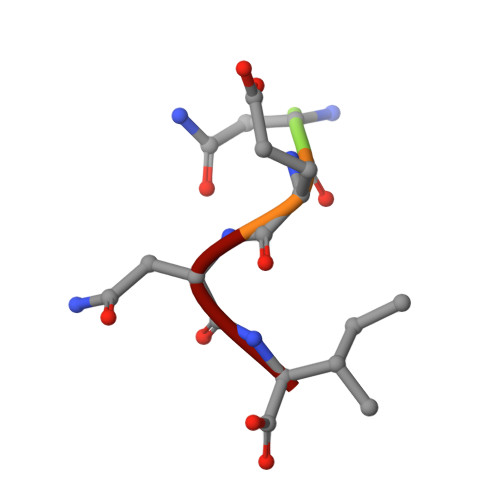
Design of o-acetylserine sulfhydrylase inhibitors by mimicking nature.
Salsi, E., Bayden, A.S., Spyrakis, F., Amadasi, A., Campanini, B., Bettati, S., Dodatko, T., Cozzini, P., Kellogg, G.E., Cook, P.F., Roderick, S.L., Mozzarelli, A.(2010) J Med Chem 53: 345-356
- PubMed: 19928859 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1021/jm901325e
- Primary Citation Related Structures:
3IQG, 3IQH, 3IQI - PubMed Abstract:
The inhibition of cysteine biosynthesis in prokaryotes and protozoa has been proposed to be relevant for the development of antibiotics. Haemophilus influenzae O-acetylserine sulfhydrylase (OASS), catalyzing l-cysteine formation, is inhibited by the insertion of the C-terminal pentapeptide (MNLNI) of serine acetyltransferase into the active site. Four-hundred MNXXI pentapeptides were generated in silico, docked into OASS active site using GOLD, and scored with HINT. The terminal P5 Ile accounts for about 50% of the binding energy. Glu or Asp at position P4 and, to a lesser extent, at position P3 also significantly contribute to the binding interaction. The predicted affinity of 14 selected pentapeptides correlated well with the experimentally determined dissociation constants. The X-ray structure of three high affinity pentapeptide-OASS complexes were compared with the docked poses. These results, combined with a GRID analysis of the active site, allowed us to define a pharmacophoric scaffold for the design of peptidomimetic inhibitors.
- Department of Biochemistry and Molecular Biology, University of Parma, Italy. andrea.mozzarelli@unipr.it
Organizational Affiliation: