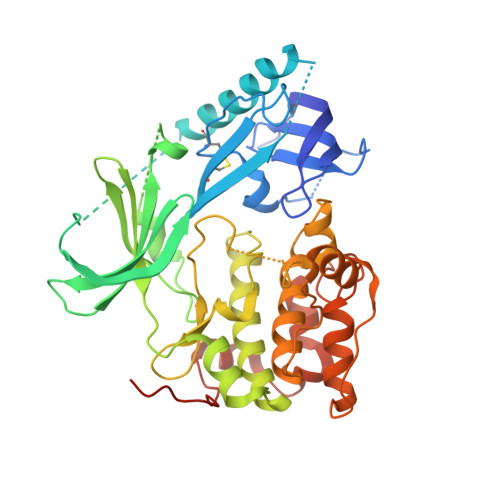
Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition.
Wu, W.I., Voegtli, W.C., Sturgis, H.L., Dizon, F.P., Vigers, G.P., Brandhuber, B.J.(2010) PLoS One 5: 12913-12913
- PubMed: 20886116 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1371/journal.pone.0012913
- Primary Citation Related Structures:
3O96 - PubMed Abstract:
AKT1 (NP_005154.2) is a member of the serine/threonine AGC protein kinase family involved in cellular metabolism, growth, proliferation and survival. The three human AKT isozymes are highly homologous multi-domain proteins with both overlapping and distinct cellular functions. Dysregulation of the AKT pathway has been identified in multiple human cancers. Several clinical trials are in progress to test the efficacy of AKT pathway inhibitors in treating cancer. Recently, a series of AKT isozyme-selective allosteric inhibitors have been reported. They require the presence of both the pleckstrin-homology (PH) and kinase domains of AKT, but their binding mode has not yet been elucidated. We present here a 2.7 Å resolution co-crystal structure of human AKT1 containing both the PH and kinase domains with a selective allosteric inhibitor bound in the interface. The structure reveals the interactions between the PH and kinase domains, as well as the critical amino residues that mediate binding of the inhibitor to AKT1. Our work also reveals an intricate balance in the enzymatic regulation of AKT, where the PH domain appears to lock the kinase in an inactive conformation and the kinase domain disrupts the phospholipid binding site of the PH domain. This information advances our knowledge in AKT1 structure and regulation, thereby providing a structural foundation for interpreting the effects of different classes of AKT inhibitors and designing selective ones.
- Department of Structural Biology, Array BioPharma Inc., Boulder, Colorado, United States of America.
Organizational Affiliation: