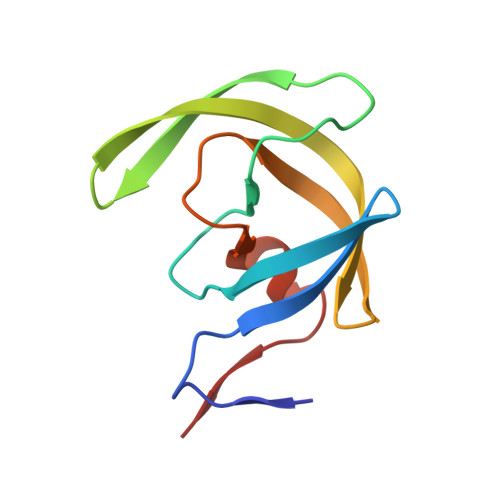
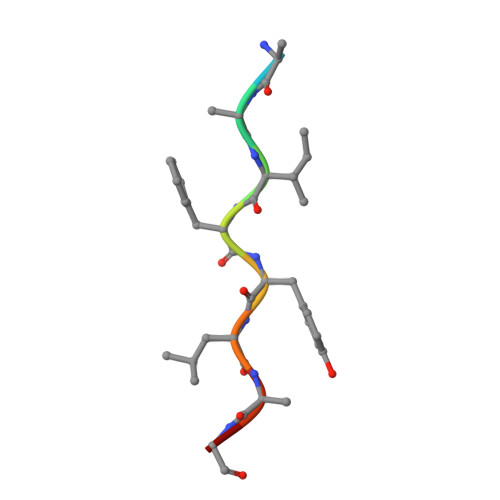
Computational design and experimental study of tighter binding peptides to an inactivated mutant of HIV-1 protease
Altman, M.D., Nalivaika, E.A., Prabu-Jeyabalan, M., Schiffer, C.A., Tidor, B.(2007) Proteins 70: 678-694
- PubMed: 17729291 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1002/prot.21514
- Primary Citation Related Structures:
2NXD, 2NXL, 2NXM - PubMed Abstract:
Drug resistance in HIV-1 protease, a barrier to effective treatment, is generally caused by mutations in the enzyme that disrupt inhibitor binding but still allow for substrate processing. Structural studies with mutant, inactive enzyme, have provided detailed information regarding how the substrates bind to the protease yet avoid resistance mutations; insights obtained inform the development of next generation therapeutics. Although structures have been obtained of complexes between substrate peptide and inactivated (D25N) protease, thermodynamic studies of peptide binding have been challenging due to low affinity. Peptides that bind tighter to the inactivated protease than the natural substrates would be valuable for thermodynamic studies as well as to explore whether the structural envelope observed for substrate peptides is a function of weak binding. Here, two computational methods-namely, charge optimization and protein design-were applied to identify peptide sequences predicted to have higher binding affinity to the inactivated protease, starting from an RT-RH derived substrate peptide. Of the candidate designed peptides, three were tested for binding with isothermal titration calorimetry, with one, containing a single threonine to valine substitution, measured to have more than a 10-fold improvement over the tightest binding natural substrate. Crystal structures were also obtained for the same three designed peptide complexes; they show good agreement with computational prediction. Thermodynamic studies show that binding is entropically driven, more so for designed affinity enhanced variants than for the starting substrate. Structural studies show strong similarities between natural and tighter-binding designed peptide complexes, which may have implications in understanding the molecular mechanisms of drug resistance in HIV-1 protease.
- Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA.
Organizational Affiliation: