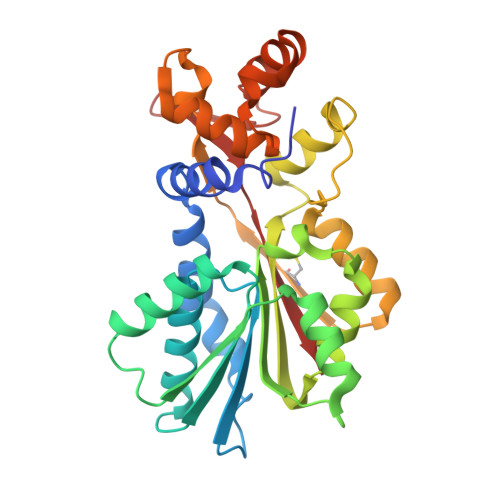
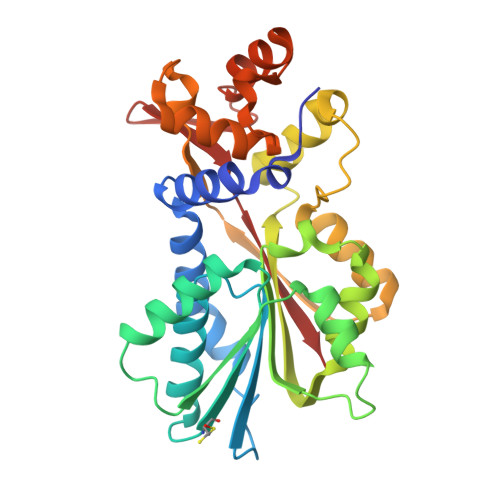
Structural basis for inhibition of histamine N-methyltransferase by diverse drugs
Horton, J.R., Sawada, K., Nishibori, M., Cheng, X.(2005) J Mol Biol 353: 334-344
- PubMed: 16168438 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1016/j.jmb.2005.08.040
- Primary Citation Related Structures:
2AOT, 2AOU, 2AOV, 2AOW, 2AOX - PubMed Abstract:
In mammals, histamine action is terminated through metabolic inactivation by histamine N-methyltransferase (HNMT) and diamine oxidase. In addition to three well-studied pharmacological functions, smooth muscle contraction, increased vascular permeability, and stimulation of gastric acid secretion, histamine plays important roles in neurotransmission, immunomodulation, and regulation of cell proliferation. The histamine receptor H1 antagonist diphenhydramine, the antimalarial drug amodiaquine, the antifolate drug metoprine, and the anticholinesterase drug tacrine (an early drug for Alzheimer's disease) are surprisingly all potent HNMT inhibitors, having inhibition constants in the range of 10-100nM. We have determined the structural mode of interaction of these four inhibitors with HNMT. Despite their structural diversity, they all occupy the histamine-binding site, thus blocking access to the enzyme's active site. Near the N terminus of HNMT, several aromatic residues (Phe9, Tyr15, and Phe19) adopt different rotamer conformations or become disordered in the enzyme-inhibitor complexes, accommodating the diverse, rigid hydrophobic groups of the inhibitors. The maximized shape complementarity between the protein aromatic side-chains and aromatic ring(s) of the inhibitors are responsible for the tight binding of these varied inhibitors.
- Department of Biochemistry Emory University School of Medicine, 1510 Clifton Road Atlanta, GA 30322, USA.
Organizational Affiliation: