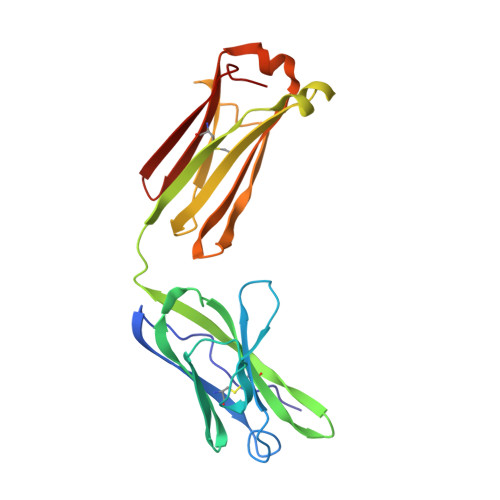
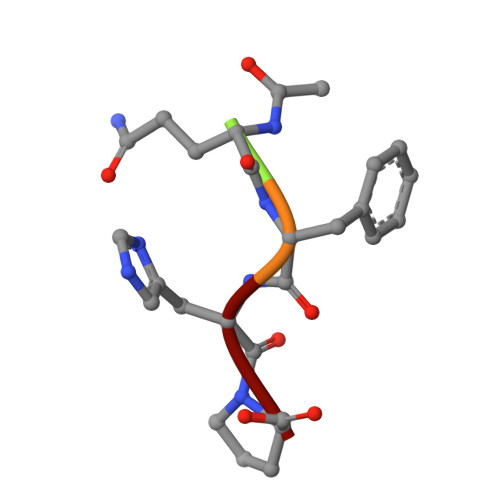
Principles and pitfalls in designing site-directed peptide ligands.
Edmundson, A.B., Harris, D.L., Fan, Z.C., Guddat, L.W., Schley, B.T., Hanson, B.L., Tribbick, G., Geysen, H.M.(1993) Proteins 16: 246-267
- PubMed: 8346191 Search on PubMed
- DOI: https://doi.org/10.1002/prot.340160304
- Primary Citation Related Structures:
1MCB, 1MCC, 1MCD, 1MCE, 1MCF, 1MCH, 1MCI, 1MCJ, 1MCK, 1MCL, 1MCN, 1MCQ, 1MCR, 1MCS - PubMed Abstract:
An immunoglobulin light chain dimer with a large generic binding cavity was used as a host molecule for designing a series of peptide guest ligands. In a screening procedure peptides coupled to solid supports were systematically tested for binding activity by enzyme linked immunosorbent assays (ELISA). Key members of the binding series were synthesized in milligram quantities and diffused into crystals of the host molecule for X-ray analyses. These peptides were incrementally increased in size and affinity until they nearly filled the cavity. Progressive changes in binding patterns were mapped by comparisons of crystallographically refined structures of 14 peptide-protein complexes at 2.7 A resolution. These comparisons led to guidelines for ligand design and also suggested ways to modify previously established binding patterns. By manipulating equilibria involving histidine, for example, it was possible to abolish one important intramolecular interaction of the bound ligand and substitute another. These events triggered a change in conformation of the ligand from a compact to an extended form and a comprehensive change in the mode of binding to the protein. In dipeptides of histidine and proline, protonation of both imidazolium nitrogen atoms was used to program an end-to-end reversal of the direction in which the ligand was inserted into the binding cavity. Peptides cocrystallized with proteins produced complexes somewhat different in structure from those in which ligands were diffused into preexisting crystals. In such a large and malleable cavity, space utilization was thus different when a ligand was introduced before the imposition of crystal packing restraints.
- Harrington Cancer Center, Amarillo, Texas 79106.
Organizational Affiliation: