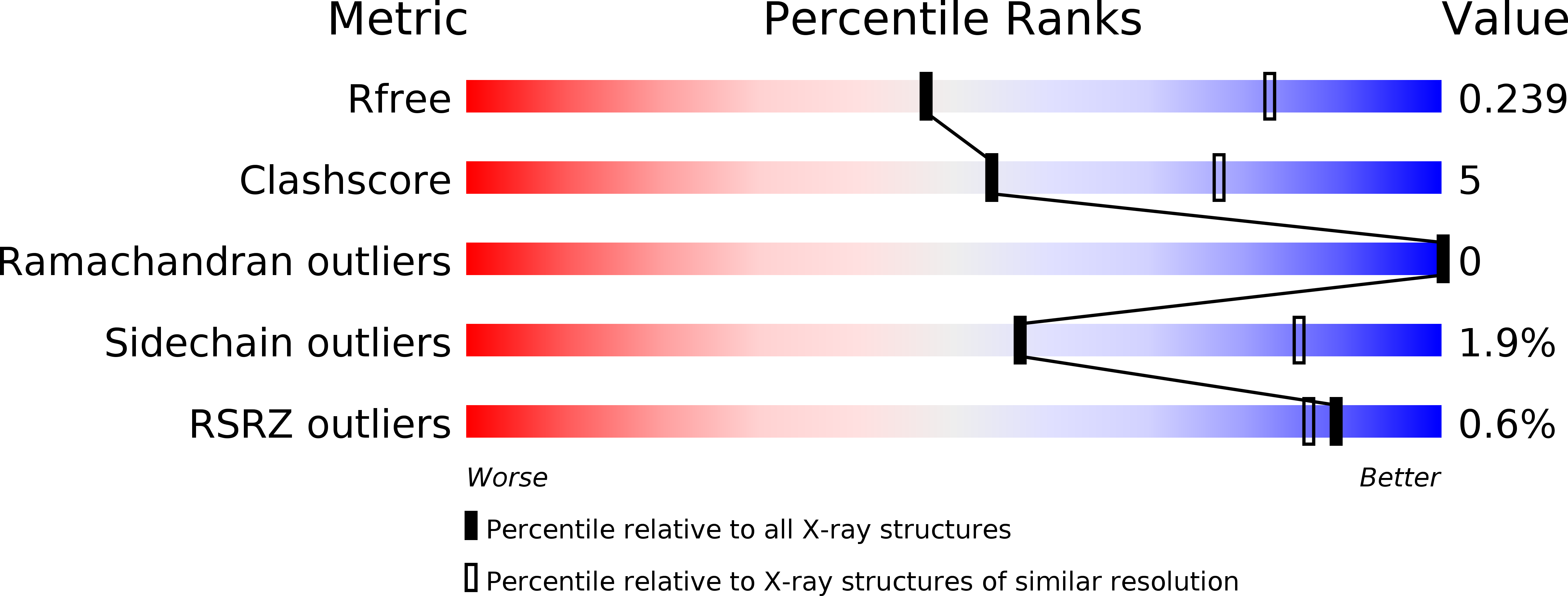
Structure of the Proteus vulgaris HigB-(HigA)2-HigB Toxin-Antitoxin Complex.
Schureck, M.A., Maehigashi, T., Miles, S.J., Marquez, J., Cho, S.E., Erdman, R., Dunham, C.M.(2014) J Biol Chem 289: 1060-1070
- PubMed: 24257752
- DOI: https://doi.org/10.1074/jbc.M113.512095
- Primary Citation of Related Structures:
4MCT, 4MCX - PubMed Abstract:
Bacterial toxin-antitoxin (TA) systems regulate key cellular processes to promote cell survival during periods of stress. During steady-state cell growth, antitoxins typically interact with their cognate toxins to inhibit activity presumably by preventing substrate recognition. We solved two x-ray crystal structures of the Proteus vulgaris tetrameric HigB-(HigA)2-HigB TA complex and found that, unlike most other TA systems, the antitoxin HigA makes minimal interactions with toxin HigB. HigB adopts a RelE family tertiary fold containing a highly conserved concave surface where we predict its active site is located. HigA does not cover the solvent-exposed HigB active site, suggesting that, in general, toxin inhibition is not solely mediated by active site hindrance by its antitoxin. Each HigA monomer contains a helix-turn-helix motif that binds to its own DNA operator to repress transcription during normal cellular growth. This is distinct from antitoxins belonging to other superfamilies that typically only form DNA-binding motifs upon dimerization. We further show that disruption of the HigB-(HigA)2-HigB tetramer to a HigBA heterodimer ablates operator binding. Taken together, our biochemical and structural studies elucidate the novel molecular details of the HigBA TA system.
Organizational Affiliation:
From the Department of Biochemistry, Emory University School of Medicine, Atlanta, Georgia 30322.