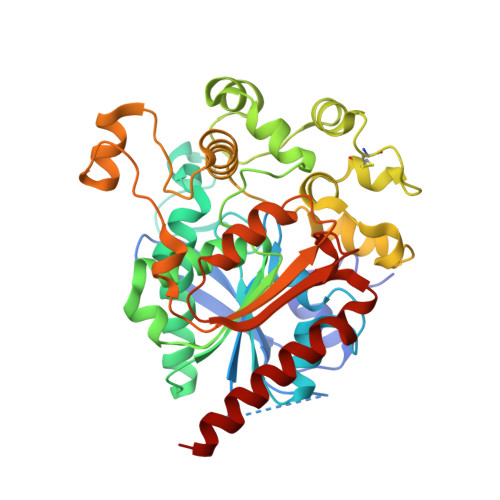
Crystal structure of human lysosomal acid lipase and its implications in cholesteryl ester storage disease.
Rajamohan, F., Reyes, A.R., Tu, M., Nedoma, N.L., Hoth, L.R., Schwaid, A.G., Kurumbail, R.G., Ward, J., Han, S.(2020) J Lipid Res 61: 1192-1202
- PubMed: 32482718 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1194/jlr.RA120000748
- Primary Citation Related Structures:
6V7N - PubMed Abstract:
Lysosomal acid lipase (LAL) is a serine hydrolase that hydrolyzes cholesteryl ester (CE) and TGs delivered to the lysosomes into free cholesterol and fatty acids. LAL deficiency due to mutations in the LAL gene ( LIPA ) results in accumulation of TGs and cholesterol esters in various tissues of the body leading to pathological conditions such as Wolman's disease and CE storage disease (CESD). Here, we present the first crystal structure of recombinant human LAL (HLAL) to 2.6 Å resolution in its closed form. The crystal structure was enabled by mutating three of the six potential glycosylation sites. The overall structure of HLAL closely resembles that of the evolutionarily related human gastric lipase (HGL). It consists of a core domain belonging to the classical α/β hydrolase-fold family with a classical catalytic triad (Ser-153, His-353, Asp-324), an oxyanion hole, and a "cap" domain, which regulates substrate entry to the catalytic site. Most significant structural differences between HLAL and HGL exist at the lid region. Deletion of the short helix, 238 NLCFLLC 244 , at the lid region implied a possible role in regulating the highly hydrophobic substrate binding site from self-oligomerization during interfacial activation. We also performed molecular dynamic simulations of dog gastric lipase (lid-open form) and HLAL to gain insights and speculated a possible role of the human mutant, H274Y, leading to CESD.
- Pfizer Worldwide Research, Groton, CT 06340 francis.rajamohan@pfizer.com.
Organizational Affiliation: