Computational Analysis of Energy Landscapes Reveals Dynamic Features That Contribute to Binding of Inhibitors to CFTR-Associated Ligand.
Holt, G.T., Jou, J.D., Gill, N.P., Lowegard, A.U., Martin, J.W., Madden, D.R., Donald, B.R.(2019) J Phys Chem B 123: 10441-10455
- PubMed: 31697075 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1021/acs.jpcb.9b07278
- Primary Citation Related Structures:
6OV7 - PubMed Abstract:
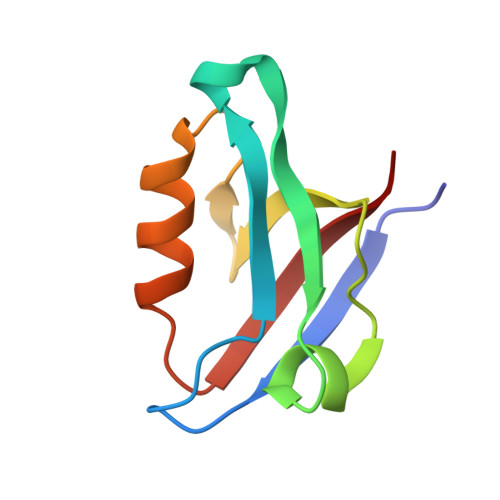
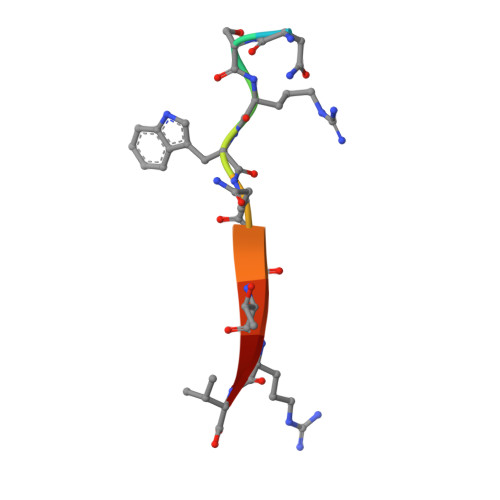
The CFTR-associated ligand PDZ domain (CALP) binds to the cystic fibrosis transmembrane conductance regulator (CFTR) and mediates lysosomal degradation of mature CFTR. Inhibition of this interaction has been explored as a therapeutic avenue for cystic fibrosis. Previously, we reported the ensemble-based computational design of a novel peptide inhibitor of CALP, which resulted in the most binding-efficient inhibitor to date. This inhibitor, kCAL01, was designed using osprey and evinced significant biological activity in in vitro cell-based assays. Here, we report a crystal structure of kCAL01 bound to CALP and compare structural features against iCAL36, a previously developed inhibitor of CALP. We compute side-chain energy landscapes for each structure to not only enable approximation of binding thermodynamics but also reveal ensemble features that contribute to the comparatively efficient binding of kCAL01. Finally, we compare the previously reported design ensemble for kCAL01 vs the new crystal structure and show that, despite small differences between the design model and crystal structure, significant biophysical features that enhance inhibitor binding are captured in the design ensemble. This suggests not only that ensemble-based design captured thermodynamically significant features observed in vitro , but also that a design eschewing ensembles would miss the kCAL01 sequence entirely.
- Department of Computer Science , Duke University , Durham , North Carolina 27708 , United States.
Organizational Affiliation: