From a novel HTS hit to potent, selective, and orally bioavailable KDM5 inhibitors.
Liang, J., Labadie, S., Zhang, B., Ortwine, D.F., Patel, S., Vinogradova, M., Kiefer, J.R., Mauer, T., Gehling, V.S., Harmange, J.C., Cummings, R., Lai, T., Liao, J., Zheng, X., Liu, Y., Gustafson, A., Van der Porten, E., Mao, W., Liederer, B.M., Deshmukh, G., An, L., Ran, Y., Classon, M., Trojer, P., Dragovich, P.S., Murray, L.(2017) Bioorg Med Chem Lett 27: 2974-2981
- PubMed: 28512031 Search on PubMed
- DOI: https://doi.org/10.1016/j.bmcl.2017.05.016
- Primary Citation Related Structures:
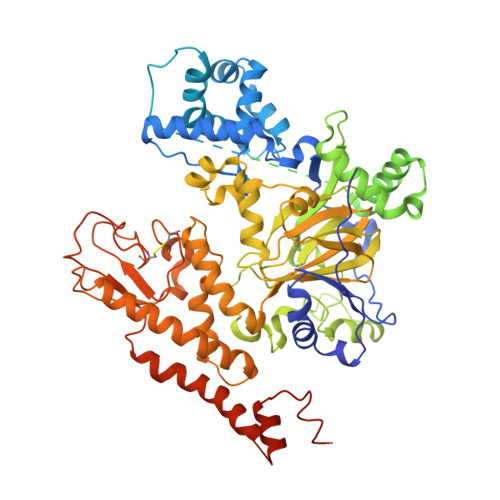
5V9P, 5V9T - PubMed Abstract:
A high-throughput screening (HTS) of the Genentech/Roche library identified a novel, uncharged scaffold as a KDM5A inhibitor. Lacking insight into the binding mode, initial attempts to improve inhibitor potency failed to improve potency, and synthesis of analogs was further hampered by the presence of a C-C bond between the pyrrolidine and pyridine. Replacing this with a C-N bond significantly simplified synthesis, yielding pyrazole analog 35, of which we obtained a co-crystal structure with KDM5A. Using structure-based design approach, we identified 50 with improved biochemical, cell potency and reduced MW and lower lipophilicity (LogD) compared with the original hit. Furthermore, 50 showed lower clearance than 9 in mice. In combination with its remarkably low plasma protein binding (PPB) in mice (40%), oral dosing of 50 at 5mg/kg resulted in unbound C max ∼2-fold of its cell potency (PC9 H3K4Me3 0.96μM), meeting our criteria for an in vivo tool compound from a new scaffold.
- Genentech Inc., 1 DNA Way, South San Francisco, CA 94080, USA. Electronic address: liang.jun@gene.com.
Organizational Affiliation: