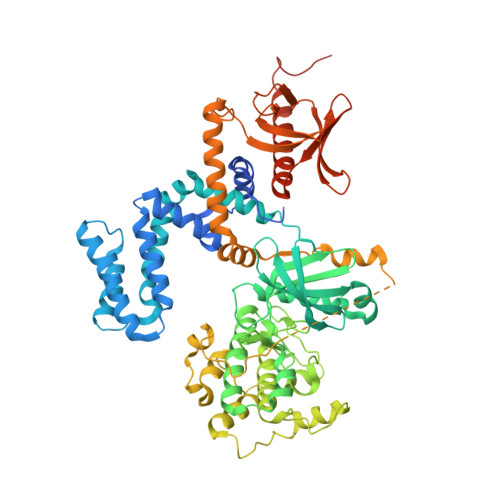
Molecular Mechanism of Selectivity among G Protein-Coupled Receptor Kinase 2 Inhibitors.
Thal, D.M., Yeow, R.Y., Schoenau, C., Huber, J., Tesmer, J.J.(2011) Mol Pharmacol 80: 294-303
- PubMed: 21596927 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1124/mol.111.071522
- Primary Citation Related Structures:
3PSC, 3PVU, 3PVW - PubMed Abstract:
G protein-coupled receptors (GPCRs) are key regulators of cell physiology and control processes ranging from glucose homeostasis to contractility of the heart. A major mechanism for the desensitization of activated GPCRs is their phosphorylation by GPCR kinases (GRKs). Overexpression of GRK2 is strongly linked to heart failure, and GRK2 has long been considered a pharmaceutical target for the treatment of cardiovascular disease. Several lead compounds developed by Takeda Pharmaceuticals show high selectivity for GRK2 and therapeutic potential for the treatment of heart failure. To understand how these drugs achieve their selectivity, we determined crystal structures of the bovine GRK2-Gβγ complex in the presence of two of these inhibitors. Comparison with the apoGRK2-Gβγ structure demonstrates that the compounds bind in the kinase active site in a manner similar to that of the AGC kinase inhibitor balanol. Both balanol and the Takeda compounds induce a slight closure of the kinase domain, the degree of which correlates with the potencies of the inhibitors. Based on our crystal structures and homology modeling, we identified five amino acids surrounding the inhibitor binding site that we hypothesized could contribute to inhibitor selectivity. However, our results indicate that these residues are not major determinants of selectivity among GRK subfamilies. Rather, selectivity is achieved by the stabilization of a unique inactive conformation of the GRK2 kinase domain.
- Life Sciences Institute, University of Michigan, 210 Washtenaw Ave., Room 3425, Ann Arbor, MI 48109, USA. tesmerjj@umich.edu
Organizational Affiliation: