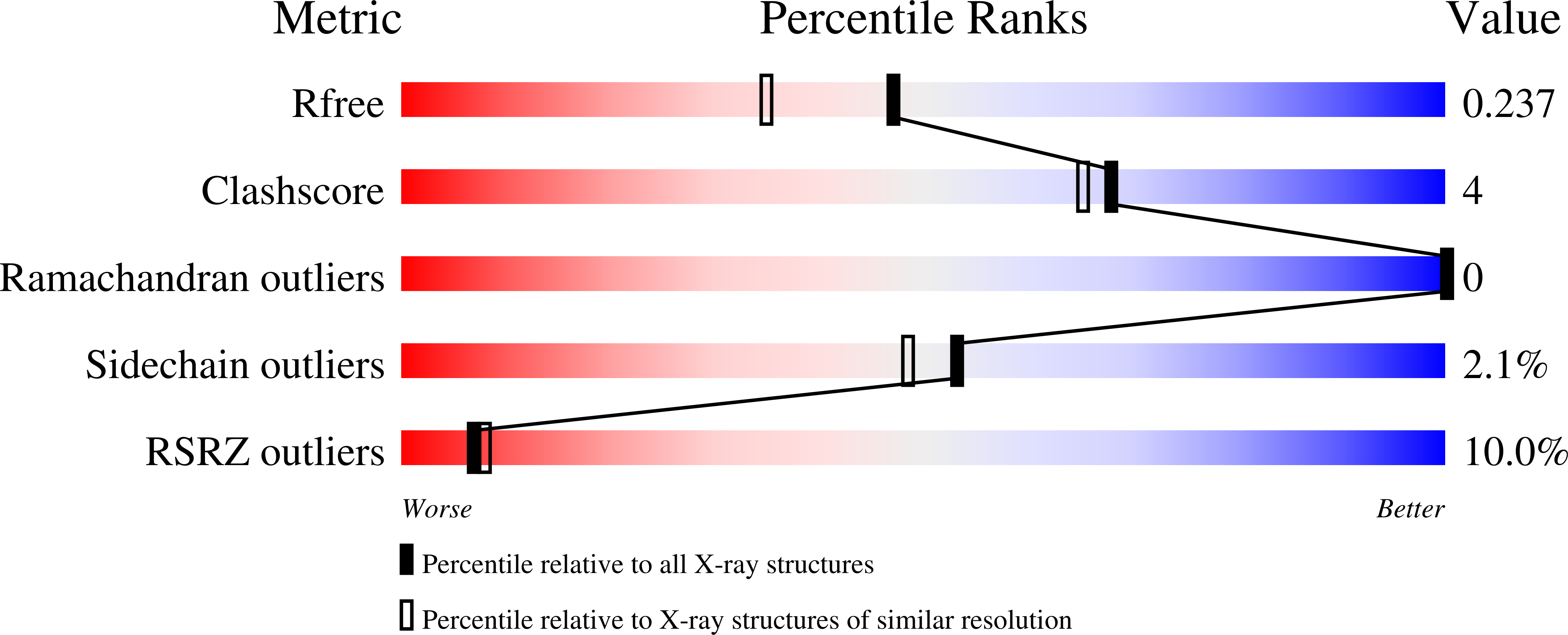
Structure-based approach to the identification of a novel group of selective glucosamine analogue inhibitors of Trypanosoma cruzi glucokinase.
D'Antonio, E.L., Deinema, M.S., Kearns, S.P., Frey, T.A., Tanghe, S., Perry, K., Roy, T.A., Gracz, H.S., Rodriguez, A., D'Antonio, J.(2016) Mol Biochem Parasitol 204: 64-76
- PubMed: 26778112
- DOI: https://doi.org/10.1016/j.molbiopara.2015.12.004
- Primary Citation of Related Structures:
5BRD, 5BRE, 5BRF, 5BRH - PubMed Abstract:
Glucokinase and hexokinase from pathogenic protozoa Trypanosoma cruzi are potential drug targets for antiparasitic chemotherapy of Chagas' disease. These glucose kinases phosphorylate d-glucose with co-substrate ATP and yield glucose 6-phosphate and are involved in essential metabolic pathways, such as glycolysis and the pentose phosphate pathway. An inhibitor class was conceived that is selective for T. cruzi glucokinase (TcGlcK) using structure-based drug design involving glucosamine having a linker from the C2 amino that terminates with a hydrophobic group either being phenyl, p-hydroxyphenyl, or dioxobenzo[b]thiophenyl groups. The synthesis and characterization for two of the four compounds are presented while the other two compounds were commercially available. Four high-resolution X-ray crystal structures of TcGlcK inhibitor complexes are reported along with enzyme inhibition constants (Ki) for TcGlcK and Homo sapiens hexokinase IV (HsHxKIV). These glucosamine analogue inhibitors include three strongly selective TcGlcK inhibitors and a fourth inhibitor, benzoyl glucosamine (BENZ-GlcN), which is a similar variant exhibiting a shorter linker. Carboxybenzyl glucosamine (CBZ-GlcN) was found to be the strongest glucokinase inhibitor known to date, having a Ki of 0.71±0.05μM. Also reported are two biologically active inhibitors against in vitro T. cruzi culture that were BENZ-GlcN and CBZ-GlcN, with intracellular amastigote growth inhibition IC50 values of 16.08±0.16μM and 48.73±0.69μM, respectively. These compounds revealed little to no toxicity against mammalian NIH-3T3 fibroblasts and provide a key starting point for further drug development with this class of compound.
Organizational Affiliation:
Department of Natural Sciences, University of South Carolina Beaufort, 1 University Boulevard, Bluffton, South Carolina 29909, USA. Electronic address: edantonio@uscb.edu.