



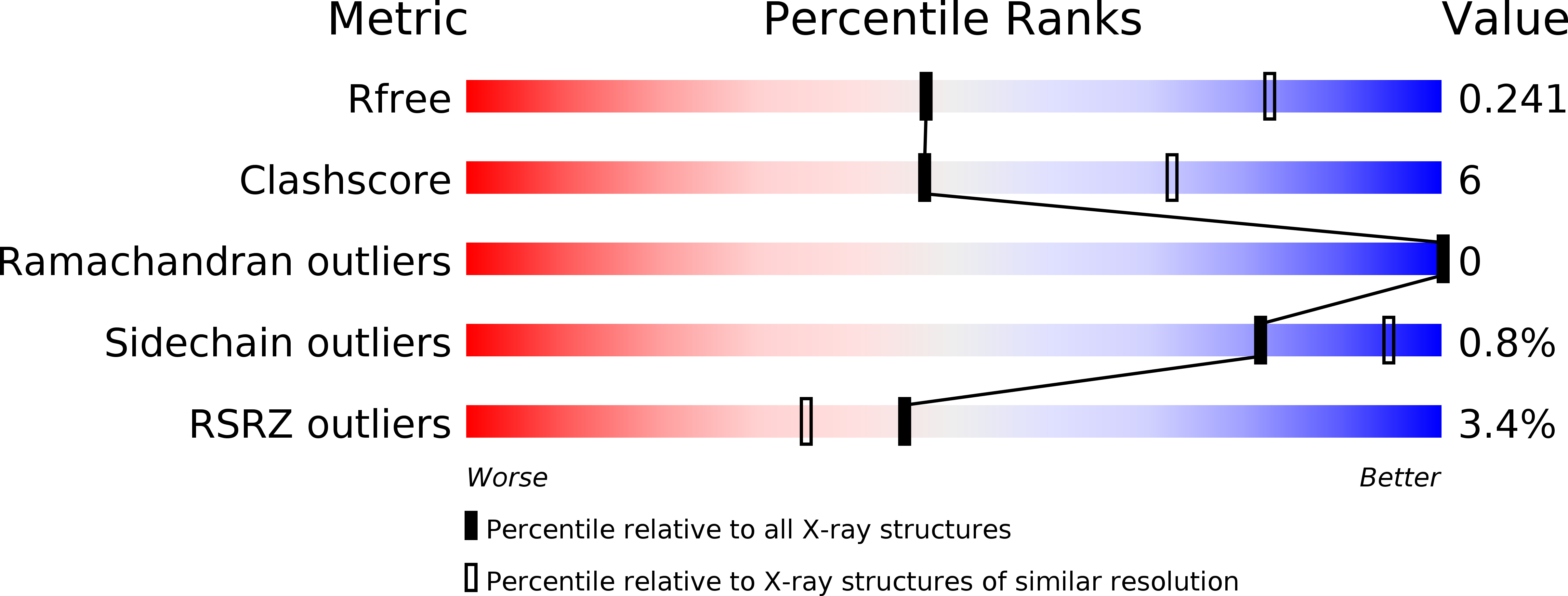
In meso in situ serial X-ray crystallography of soluble and membrane proteins.
Huang, C.Y., Olieric, V., Ma, P., Panepucci, E., Diederichs, K., Wang, M., Caffrey, M.(2015) Acta Crystallogr D Biol Crystallogr 71: 1238-1256
- PubMed: 26057665
- DOI: https://doi.org/10.1107/S1399004715005210
- Primary Citation of Related Structures:
4XJB, 4XJD, 4XJF, 4XJG, 4XJH, 4XJI, 4XNI, 4XNJ, 4XNK, 4XNL - PubMed Abstract:
The lipid cubic phase (LCP) continues to grow in popularity as a medium in which to generate crystals of membrane (and soluble) proteins for high-resolution X-ray crystallographic structure determination. To date, the PDB includes 227 records attributed to the LCP or in meso method. Among the listings are some of the highest profile membrane proteins, including the β2-adrenoreceptor-Gs protein complex that figured in the award of the 2012 Nobel Prize in Chemistry to Lefkowitz and Kobilka. The most successful in meso protocol to date uses glass sandwich crystallization plates. Despite their many advantages, glass plates are challenging to harvest crystals from. However, performing in situ X-ray diffraction measurements with these plates is not practical. Here, an alternative approach is described that provides many of the advantages of glass plates and is compatible with high-throughput in situ measurements. The novel in meso in situ serial crystallography (IMISX) method introduced here has been demonstrated with AlgE and PepT (alginate and peptide transporters, respectively) as model integral membrane proteins and with lysozyme as a test soluble protein. Structures were solved by molecular replacement and by experimental phasing using bromine SAD and native sulfur SAD methods to resolutions ranging from 1.8 to 2.8 Å using single-digit microgram quantities of protein. That sulfur SAD phasing worked is testament to the exceptional quality of the IMISX diffraction data. The IMISX method is compatible with readily available, inexpensive materials and equipment, is simple to implement and is compatible with high-throughput in situ serial data collection at macromolecular crystallography synchrotron beamlines worldwide. Because of its simplicity and effectiveness, the IMISX approach is likely to supplant existing in meso crystallization protocols. It should prove particularly attractive in the area of ligand screening for drug discovery and development.
Organizational Affiliation:
Membrane Structural and Functional Biology Group, Schools of Medicine and Biochemistry and Immunology, Trinity College, Dublin, Ireland.




