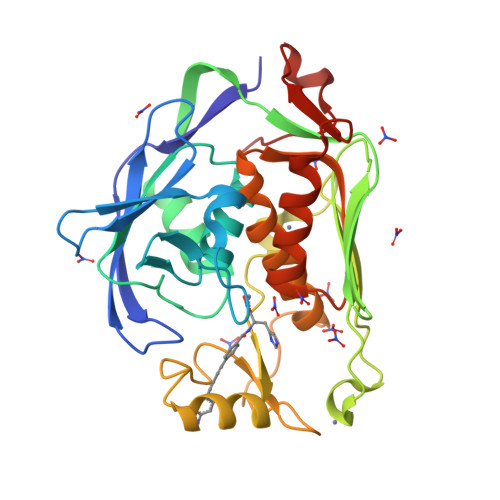
Synthesis, Structure, and Antibiotic Activity of Aryl-Substituted LpxC Inhibitors.
Liang, X., Lee, C.J., Zhao, J., Toone, E.J., Zhou, P.(2013) J Med Chem 56: 6954-6966
- PubMed: 23914798
- DOI: https://doi.org/10.1021/jm4007774
- Primary Citation of Related Structures:
4LCF, 4LCG, 4LCH - PubMed Abstract:
The zinc-dependent deacetylase LpxC catalyzes the committed step of lipid A biosynthesis in Gram-negative bacteria and is a validated target for the development of novel antibiotics to combat multidrug-resistant Gram-negative infections. Many potent LpxC inhibitors contain an essential threonyl-hydroxamate headgroup for high-affinity interaction with LpxC. We report the synthesis, antibiotic activity, and structural and enzymatic characterization of novel LpxC inhibitors containing an additional aryl group in the threonyl-hydroxamate moiety, which expands the inhibitor-binding surface in LpxC. These compounds display enhanced potency against LpxC in enzymatic assays and superior antibiotic activity against Francisella novicida in cell culture. The comparison of the antibiotic activities of these compounds against a leaky Escherichia coli strain and the wild-type strain reveals the contribution of the formidable outer-membrane permeability barrier that reduces the compounds efficacy in cell culture and emphasizes the importance of maintaining a balanced hydrophobicity and hydrophilicity profile in developing effective LpxC-targeting antibiotics.
Organizational Affiliation:
Department of Chemistry, Duke University, Durham, NC 27708, USA.