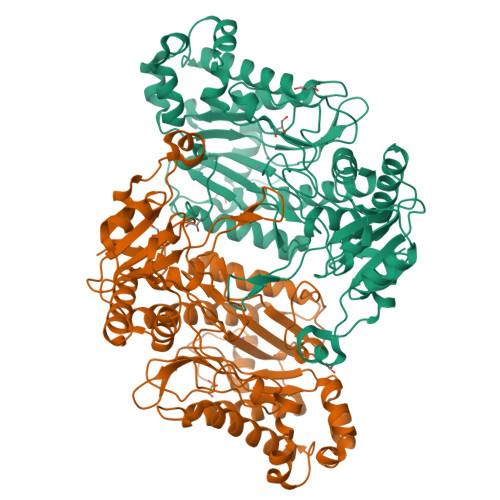
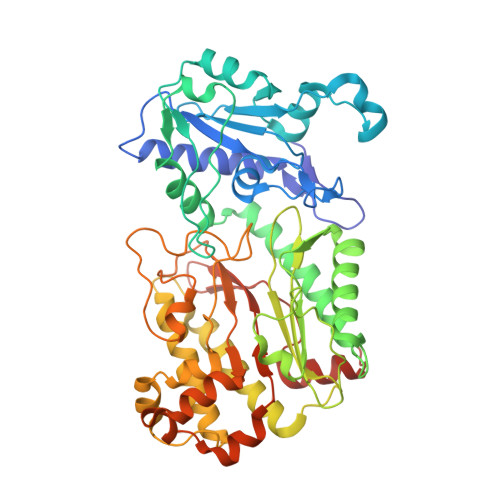
Structural analysis of new compound heterozygous variants in PEPD gene identified in a patient with Prolidase Deficiency diagnosed by exome sequencing.
Linhares, N.D., Wilk, P., Wator, E., Tostes, M.A., Weiss, M.S., Pena, S.D.J.(2021) Genet Mol Biol 44: e20200393-e20200393
- PubMed: 33877262
- DOI: https://doi.org/10.1590/1678-4685-GMB-2020-0393
- Primary Citation of Related Structures:
6QSB, 6QSC - PubMed Abstract:
Prolidase Deficiency (PD) is an autosomal recessive rare disorder caused by loss or reduction of prolidase enzymatic activity due to variants in the PEPD gene. PD clinical features vary among affected individuals: skin ulcerations, recurrent infections, and developmental delay are common. In this study, we describe a 16-year-old boy with a mild PD phenotype comprising chronic eczema, recurrent infections and elevated IgE. Whole exome sequencing analysis revealed three PEPD variants: c.575T>C p.(Leu192Pro) inherited from the mother, and c.692_694del p.(Tyr231del) and c.1409G>A p.(Arg470His), both inherited from the father. The variant p.(Tyr231del) has been previously characterized by high-resolution X-ray structure analysis as altering protein dynamics/flexibility. In order to study the effects of the other two prolidase variants, we performed site directed mutagenesis purification and crystallization studies. A high-resolution X-ray structure could only be obtained for the p.(Arg470His) variant, which showed no significant structural differences in comparison to WT prolidase. On the other hand, the p.(Leu192Pro) variant led to significant protein destabilization. Hence, we conclude that the maternal p.(Leu192Pro) variant was likely causally associated with the proband´s disease, together with the known pathogenic paternal variant p.(Tyr231del). Our results demonstrated the utility of exome sequencing to perform diagnosis in PD cases with mild phenotype.
Organizational Affiliation:
Universidade Federal de Minas Gerais, Faculdade de Medicina, Laboratório de Genômica Clínica, Belo Horizonte, MG, Brazil.