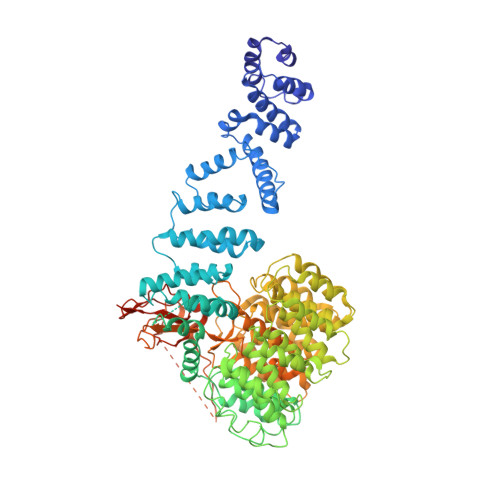
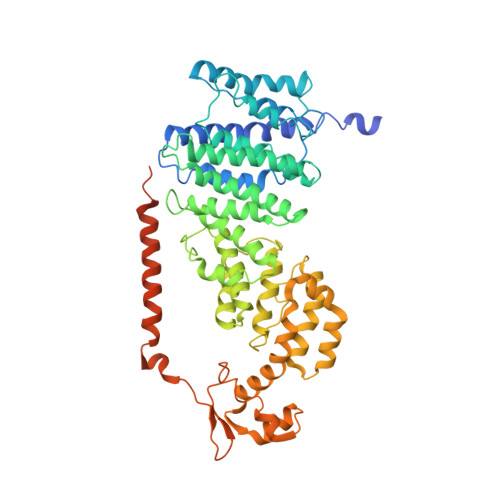
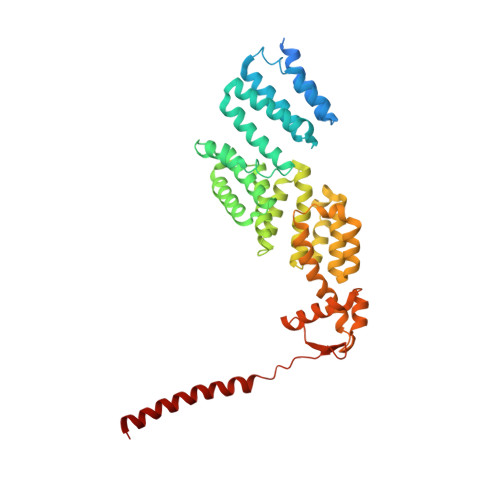
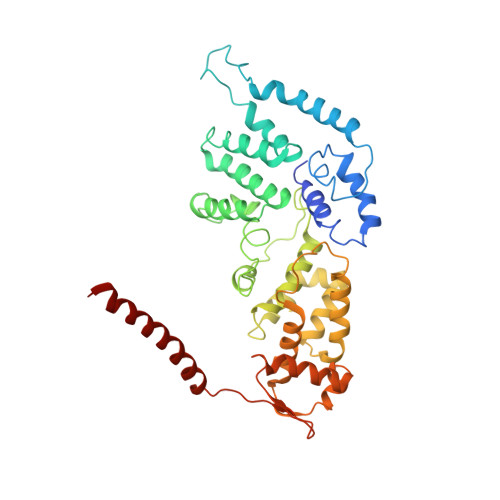
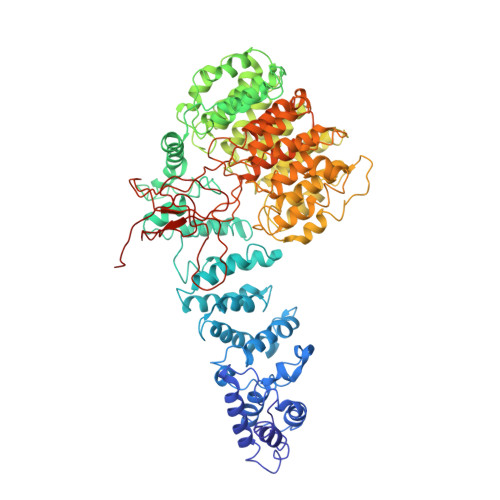
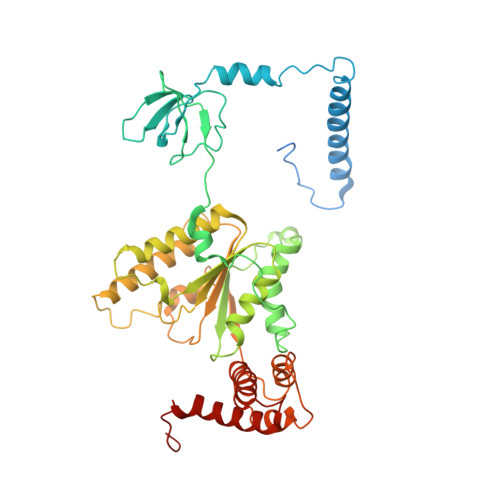
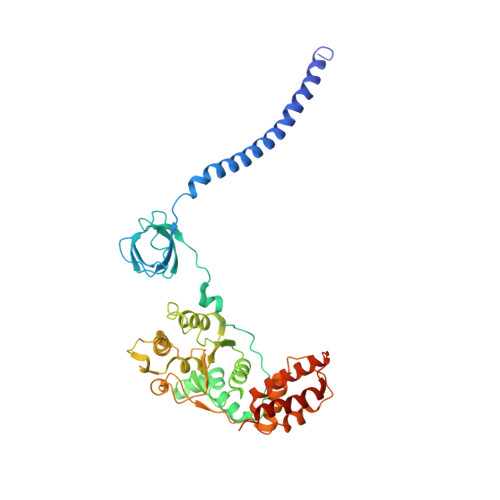
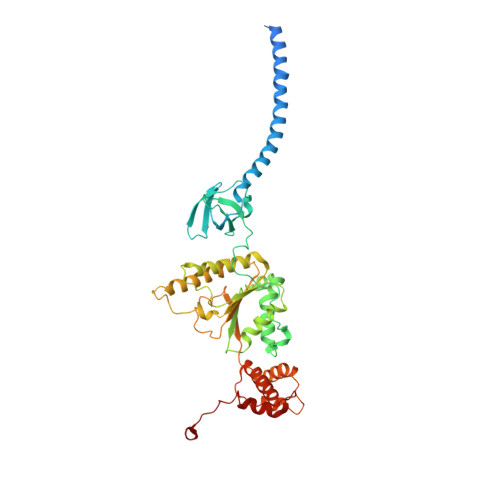
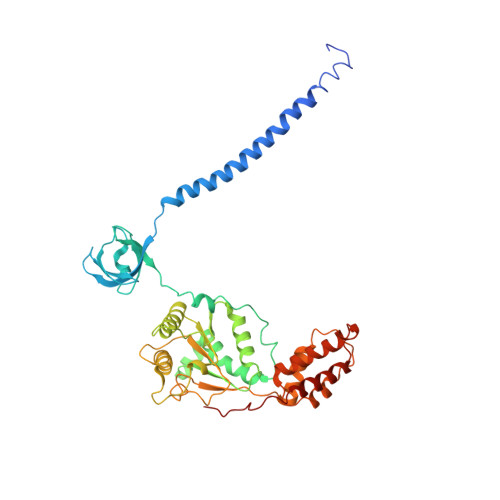
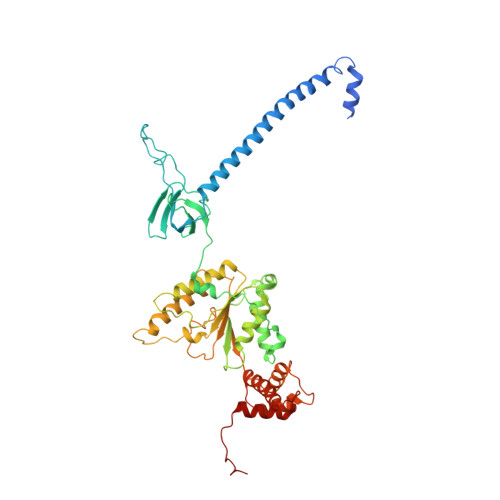
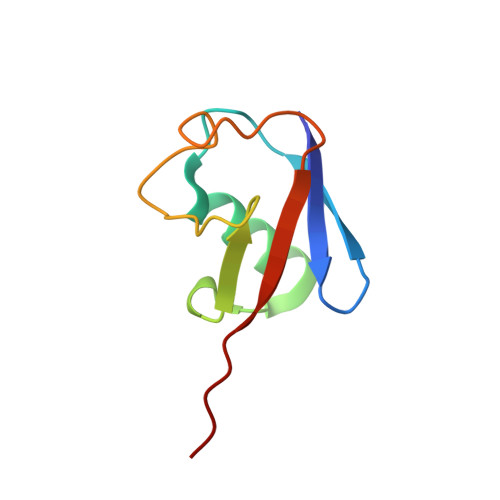
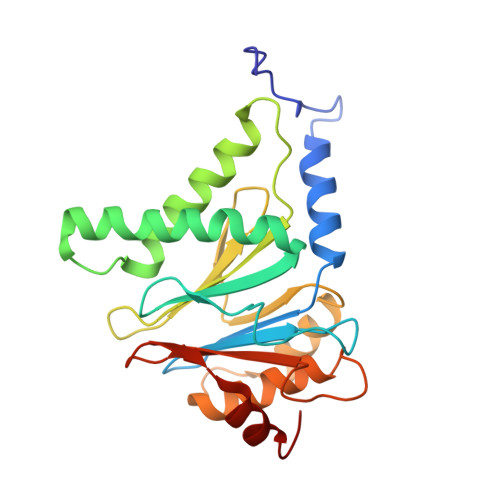
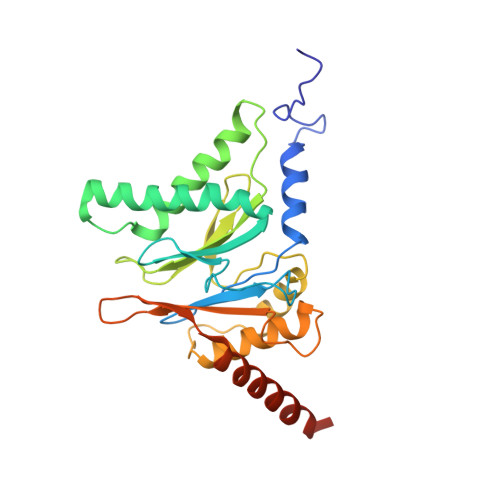
Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome.
Dong, Y., Zhang, S., Wu, Z., Li, X., Wang, W.L., Zhu, Y., Stoilova-McPhie, S., Lu, Y., Finley, D., Mao, Y.(2019) Nature 565: 49-55
- PubMed: 30479383
- DOI: https://doi.org/10.1038/s41586-018-0736-4
- Primary Citation of Related Structures:
6MSB, 6MSD, 6MSE, 6MSG, 6MSH, 6MSJ, 6MSK - PubMed Abstract:
The proteasome is an ATP-dependent, 2.5-megadalton molecular machine that is responsible for selective protein degradation in eukaryotic cells. Here we present cryo-electron microscopy structures of the substrate-engaged human proteasome in seven conformational states at 2.8-3.6 Å resolution, captured during breakdown of a polyubiquitylated protein. These structures illuminate a spatiotemporal continuum of dynamic substrate-proteasome interactions from ubiquitin recognition to substrate translocation, during which ATP hydrolysis sequentially navigates through all six ATPases. There are three principal modes of coordinated hydrolysis, featuring hydrolytic events in two oppositely positioned ATPases, in two adjacent ATPases and in one ATPase at a time. These hydrolytic modes regulate deubiquitylation, initiation of translocation and processive unfolding of substrates, respectively. Hydrolysis of ATP powers a hinge-like motion in each ATPase that regulates its substrate interaction. Synchronization of ATP binding, ADP release and ATP hydrolysis in three adjacent ATPases drives rigid-body rotations of substrate-bound ATPases that are propagated unidirectionally in the ATPase ring and unfold the substrate.
Organizational Affiliation:
State Key Laboratory for Artificial Microstructures and Mesoscopic Physics, School of Physics, Peking University, Beijing, China.