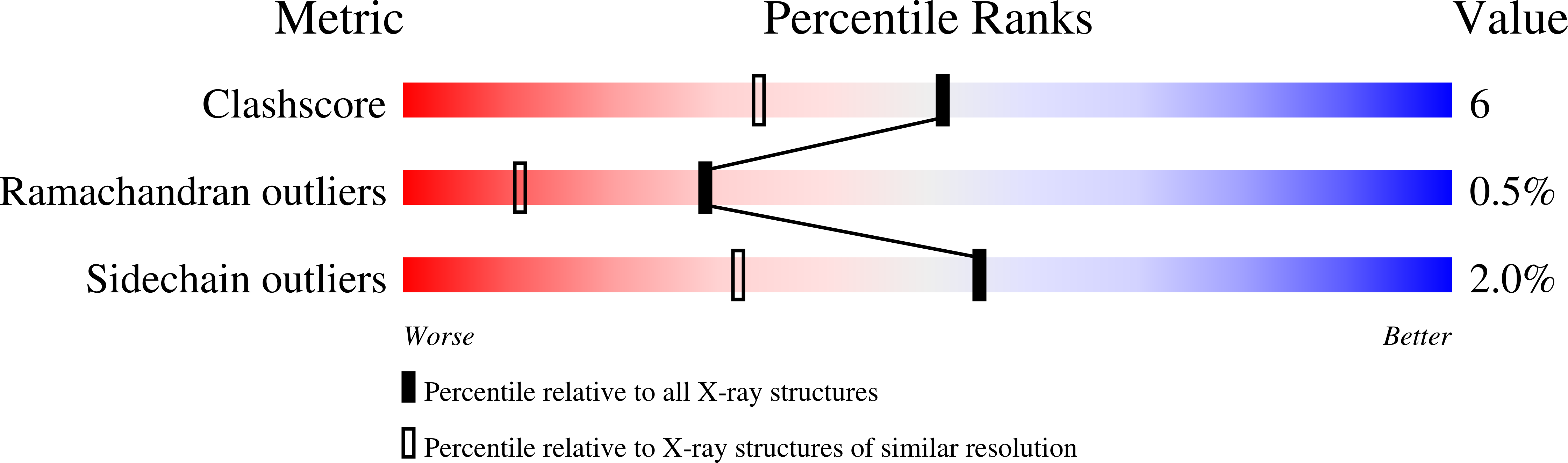
The Crystal Structure of a Complex of Campylobacter Jejuni Dutpase with Substrate Analogue Sheds Light on the Mechanism and Suggests the "Basic Module" for Dimeric D(C/U)Tpases
Moroz, O.V., Harkiolaki, M., Galperin, M.Y., Vagin, A.A., Gonzalez-Pacanowska, D., Wilson, K.S.(2004) J Mol Biol 342: 1583
- PubMed: 15364583
- DOI: https://doi.org/10.1016/j.jmb.2004.07.050
- Primary Citation of Related Structures:
1W2Y - PubMed Abstract:
The crystal structure of the dUTPase from the important gastric pathogen Campylobacter jejuni has been solved at 1.65 A spacing. This essential bacterial enzyme is the second representative of the new family of dimeric dUTPases to be structurally characterised. Members of this family have a novel all-alpha fold and are unrelated to the all-beta dUTPases of the majority of organisms including eukaryotes such as humans, bacteria such as Escherichia coli, archaea like Methanococcus jannaschii and animal viruses. Therefore, dimeric dUTPases can be considered as candidate drug targets. The X-ray structure of the C.jejuni dUTPase in complex with the non-hydrolysable substrate analogue dUpNHp allows us to define the positions of three catalytically significant phosphate-binding magnesium ions and provides a starting point for a detailed understanding of the mechanism of dUTP/dUDP hydrolysis by dimeric dUTPases. Indeed, a water molecule present in the structure is ideally situated to act as the attacking nucleophile during hydrolysis. A comparison of the dUTPases from C.jejuni and Trypanosoma cruzi reveals a common fold with certain distinct features, both in the rigid and mobile domains as defined in the T.cruzi structure. Homologues of the C.jejuni dUTPase have been identified in several other bacteria and bacteriophages, including the dCTPase of phage T4. Sequence comparisons of these proteins define a new superfamily of d(C/U)TPases that includes three distinct enzyme families: (1) dUTPases in trypanosomatides, C.jejuni and several other Gram-negative bacteria, (2) predicted dUTPases in various Gram-positive bacteria and their phages, and (3) dCTP/dUTPases in enterobacterial T4-like phages. All these enzymes share a basic module that consists of two alpha-helices from the rigid domain, two helices from the mobile domain and connecting loops. These results in concert with a number of conserved residues responsible for interdomain cross-talk provide valuable insight towards rational drug design.
Organizational Affiliation:
Structural Biology Laboratory, Department of Chemistry, University of York, Heslington, York YO10 5YW, UK.